Capecitabine is considered to have moderate potential for nausea and vomiting.
Adjuvant colon cancer: The following table shows the adverse reactions occurring in at least 5% of patients from one phase 3 trial in patients with patients with Duke stage C colon cancer who received at least 1 dose of study medication and had at least 1 safety assessment.
A total of 995 patients were treated with 1250 mg/m2 twice a day of capecitabine administered for 2 weeks followed by a 1-week rest period and 974 patients were administered 5-fluorouracil and leucovorin (20 mg/m2 leucovorin IV followed by 425 mg/m2 IV bolus 5-fluorouracil, on days 1 to 5 every 28 days). The median duration of treatment was 164 days for capecitabine treated patients and 145 days for 5-fluorouracil/leucovorin-treated patients. A total of 112 (11%) and 73 (7%) capecitabine and 5-fluorouracil/leucovorin-treated patients, respectively, discontinued treatment because of adverse reactions. A total of 18 deaths due to all causes occurred either on study or within 28 of receiving study drug: 8 (0.8%) patients randomized to capecitabine and 10 (1%) randomized to 5-fluorouracil/leucovorin. (See Tables 4 and 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Metastatic colorectal cancer: The following table shows the adverse reactions occurring in greater than or equal to 5% of patients from pooling the 2 phase 3 trials in first line metastatic colorectal cancer. A total of 596 patients with metastatic colorectal cancer were treated with 1250 mg/m
2 twice a day of capecitabine administered for 2 weeks followed by a 1-week rest period and 593 patients were administered 5-fluorouracil and leucovorin in the Mayo regimen (20 mg/m2 leucovorin IV followed by 425 mg/m2 IV bolus 5-fluorouracil, on days 1 to 5, every 28 days). In the pooled colorectal database the median duration of treatment was 139 days for capecitabine-treated patients and 140 days for 5-fluorouracil/leucovorin treated patients. A total of 78 (13%) and 63 (11%) capecitabine and 5-fluorouracil/leucovorin-treated patients, respectively, discontinued treatment because of adverse reaction/intercurrent illness. A total of 82 deaths due to all causes occurred either on study or within 28 days of receiving study drug: 50 (8.4%) patients randomized to capecitabine and 32 (5.4%) randomized to 5-fluorouracil/leucovorin. (See Table 6.)
Click on icon to see table/diagram/image
Breast cancer combination: The following data are shown for the combination study with capecitabine and docetaxel in patients with metastatic breast cancer. In the capecitabine and docetaxel combination arm the treatment was capecitabine administered orally 1250 mg/m2 twice daily as intermittent therapy (2 weeks of treatment followed by 1 week without treatment) for at least 6 weeks and docetaxel administrated as a 1-hour IV infusion at a dose of 75 mg/m2 on the first day of each 3-week cycle for at least 6 weeks. In the monotherapy arm, docetaxel was administered as a 1-hour IV infusion at a dose of 100 mg/m2 on the first day of each 3-week cycle for at least 6 weeks. The mean duration of treatment was 129 days in the combination arm and 98 days in the monotherapy arm. A total of 66 patients (26%) in the combination arm and 49 (19%) in the monotherapy arm withdrew from the study because of adverse reactions. The percentage of patients requiring dose reductions due to adverse reactions were 65% in the combination arm and 36% in the monotherapy arm. The percentage of patients requiring treatment interruptions due to adverse reactions in the combination arm was 79%. Treatment interruptions were part of the dose modification scheme for the combination therapy arm but not for the docetaxel monotherapy-treated patients. (See Table 7.)
Click on icon to see table/diagram/image
Breast cancer capecitabine monotherapy: The following data are shown for the study in stage IV breast cancer patients who received a dose of 1250 mg/m2 administered twice daily for 2 weeks followed by a 1-week rest period. The mean duration of treatment was 114 days. A total of 13 out of 62 patients (8%) discontinued treatment because of adverse reactions/intercurrent illness. (See Table 8.)
Click on icon to see table/diagram/image
Capecitabine and docetaxel in combination: Shown by body system are the clinically relevant adverse reactions in less than 5% of patients in the overall clinical trial safety database of 251 patients (study details) reported as related to the administration of capecitabine in combination with docetaxel and that were clinically at least remoted relevant. In parentheses is the incidence of grade 3 and 4 occurrence of each adverse reaction. It is anticipated that the same types of adverse reactions observed in the capecitabine monotherapy studies may be observed in patients treated with the combination of capecitabine plus docetaxel.
Cardiovascular: Hypotension (1.2%), postural hypotension (0.8%) supraventricular tachycardia (0.39%), syncope (1.2%), venous phlebitis and thrombophlebitis (0.39%).
CNS: Ataxia (0.39%), polyneuropathy (0.39%), migraine (0.39%).
GI: Ileus (0.39%), necrotizing enterocolitis (0.39%), esophageal ulcer (0.39%), hemorrhagic diarrhea (0.8%), taste loss (0.8%).
Hematologic/Lymphatic: Agranulocytosis (0.39%), prothrombin decreased (0.39%).
Hepatic: Abnormal liver function tests, hepatic coma, hepatic failure, hepatotoxic, jaundice (0.39%).
Miscellaneous: Bronchopneumonia (0.39%), hypersensitivity (1.2%), neutropenic sepsis (2.39%), renal failure (0.39), sepsis (0.39%).
Capecitabine monotherapy metastatic breast and colorectal cancer: Shown by body system are the clinically relevant adverse reactions in less than 5% patients in the overall clinical trial safety database of 875 patients (phase 3 colorectal studies 596 patients, phase 2 colorectal study 34 patients, phase 2 breast cancer studies 245 patients reported as related to the administration of capecitabine and that were clinically at least remotely relevant. In parentheses is the incidence of grade 3 or 4 occurrences of each adverse reaction.
Cardiovascular: Atrial fibrillation, bradycardia, cerebrovascular accident, extrasystoles, hypertension, myocarditis, tachycardia, ventricular extrasystoles (0.1%), hypotension, pulmonary embolism (0.2%), pericardial effusion.
CNS: Ataxia, insomnia (0.5%), confusion, depression, difficult walking, dysphasia, encephalopathy, irritability, tremor (0.1%), abnormal coordination, dysarthria, loss of consciousness (0.2%), impaired balance, sedation, vertigo.
Dermatologic: Nail disorder, photosensitivity reaction, sweating increased (0.1%), pruritus, radiation recall syndrome, skin ulceration (0.2%).
GI: Abdominal distension, ascites, dysphagia, gastric ulcer, gastroenteritis, proctalgia, toxic dilation of dilation of intestine (0.1%), ileus (0.3%).
Hematologic/Lymphatic: Leukopenia (0.2%), bone marrow depression, coagulation disorder, lymphedema, pancytopenia (0.1%), idiopathic thrombocytopenia purpura (1%).
Hepatic: Cholestatic hepatitis, hepatic fibrosis, hepatitis (0.1%), abnormal liver function tests.
Metabolic/Nutritional: Cachexia, increased weight (0.4%), hypertriglyceridemia (0.1%), edema, hypokalemia, hypomagnesemia.
Musculoskeletal: Arthritis, bone pain, myalgia (0.1%), muscle weakness.
Respiratory: Cough, epitaxis, hemoptysis, respiratory distress (0.1%), asthma, bronchitis, bronchopneumonia, pneumonia (0.2%), dyspnea.
Postmarketing: Hepatic failure, lacrimal duct stenosis.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image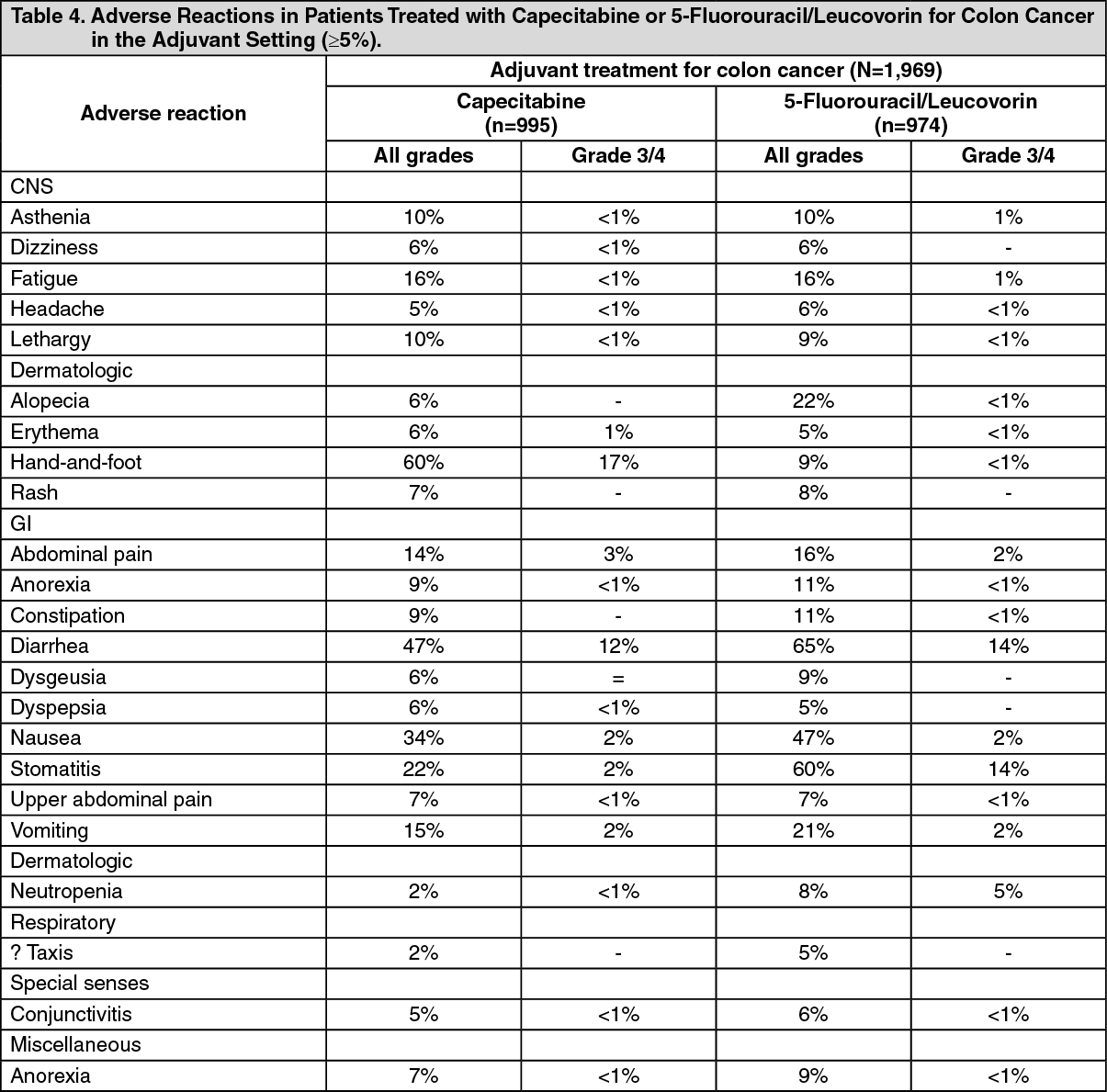 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image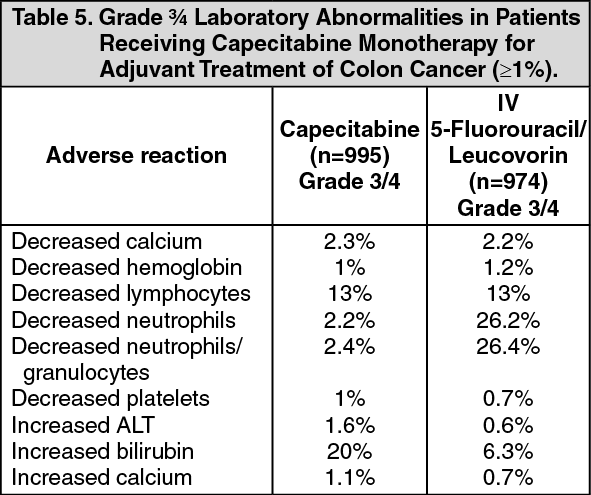 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image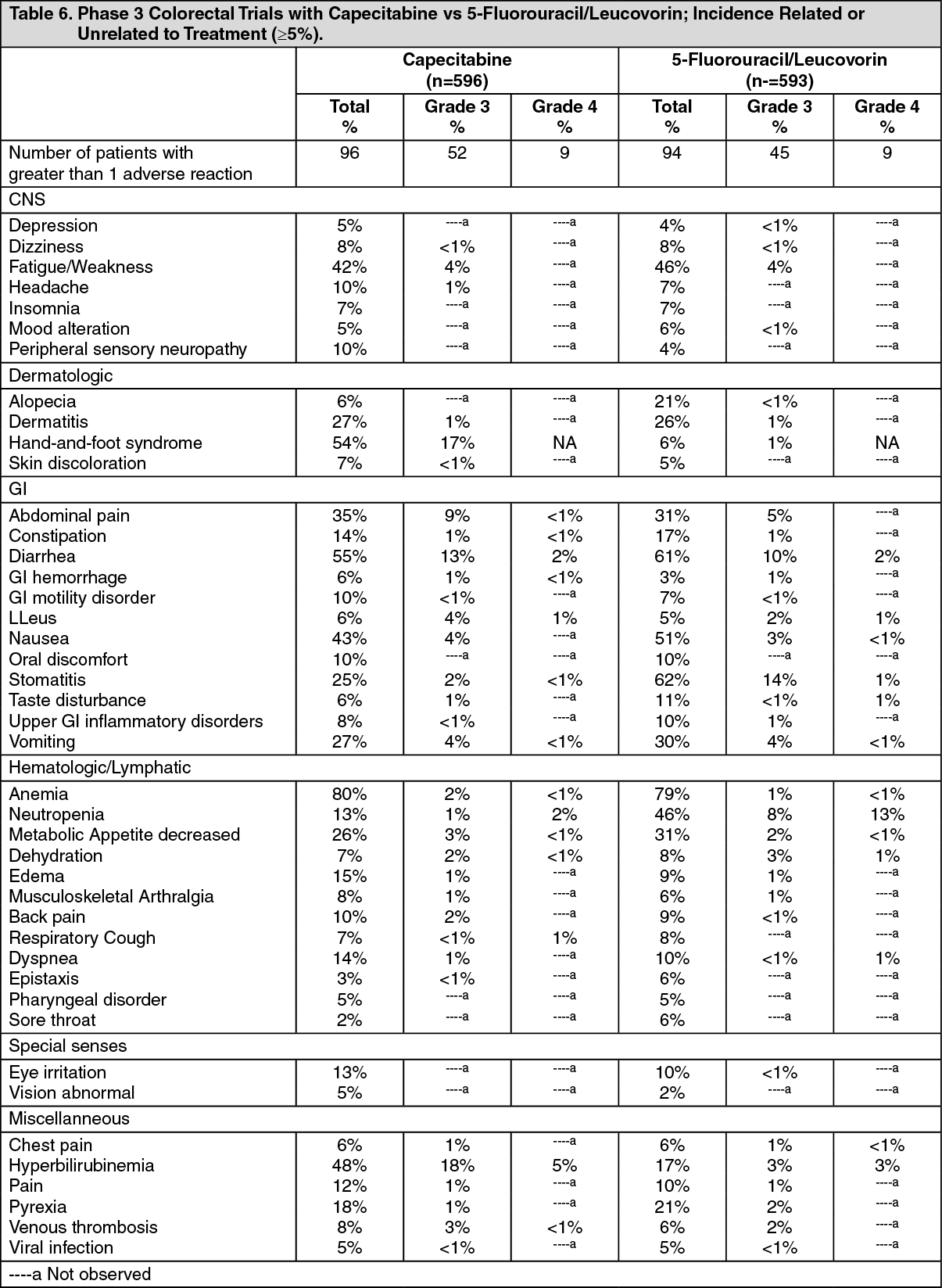 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image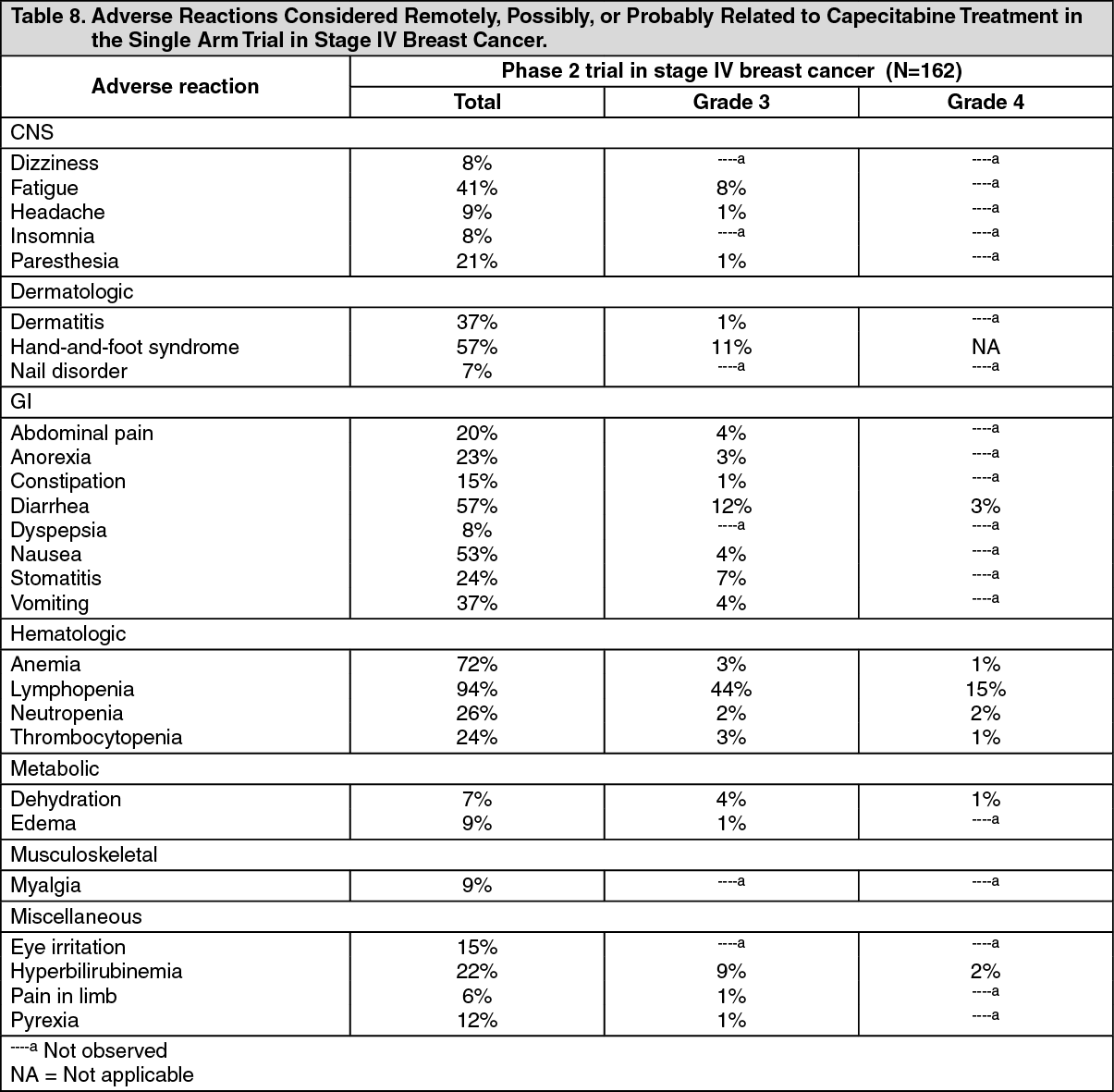 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
